Mở đầu
Công nghệ scRNA-Seq đã mở ra một hướng tiếp cận tiên tiến giúp khắc phục những hạn chế của phương pháp phân tích hệ gen phiên mã (RNA-Seq) cụm/nhóm (bulk RNA-Seq) trong nghiên cứu thực vật [1]. Các phương pháp bulk RNA-Seq thường thu được dữ liệu của toàn bộ mô hoặc cơ quan thực vật, tuy nhiên lại bỏ qua các thông tin quan trọng liên quan đến tính dị biệt phiên mã giữa các tế bào, ngay cả khi chúng cùng thuộc một loại mô. Sự khác biệt này có thể do các yếu tố nội bào như sự phân hóa tế bào, trạng thái sinh lý, chu kỳ tế bào hoặc do tác động của các yếu tố ngoại bào như ánh sáng, nhiệt độ và tín hiệu sinh hóa từ các tế bào lân cận [2]. Chẳng hạn, trong một mô lá, mô giậu có các gen liên quan đến quang hợp biểu hiện mạnh thì tế bào khí khổng bên cạnh lại kích hoạt các tín hiệu điều hòa thoát hơi nước. Công nghệ scRNA-Seq cho phép phân tích sự biểu hiện gen với độ phân giải từng tế bào, từ đó không chỉ giúp xác định chức năng của các nhóm tế bào, mà còn có thể cung cấp thông tin chi tiết hơn về các quá trình điều hòa phiên mã và hoạt động chức năng khác nhau của từng loại tế bào riêng biệt [3].
Ứng dụng của công nghệ scRNA-Seq trong nghiên cứu thực vật đã tạo ra những bước tiến đáng kể trong việc hiểu rõ hơn về sinh lý thực vật ở cấp độ tế bào. Công nghệ này đã giúp phát hiện các kiểu biểu hiện gen đặc thù của từng loại tế bào trong quá trình phát triển, đáp ứng với các yếu tố môi trường và các con đường tín hiệu phân tử quan trọng. Bên cạnh đó, việc lập bản đồ hệ gen phiên mã tế bào đơn không chỉ cho phép xác định chức năng của từng loại tế bào, mà còn cung cấp thông tin về sự tương tác giữa các tế bào trong mô, từ đó làm sáng tỏ các cơ chế điều hòa sinh trưởng và phát triển của thực vật [2]. Trong những năm gần đây, scRNA-Seq đã được ứng dụng rộng rãi để nghiên cứu phản ứng của thực vật với các điều kiện bất thuận, như hạn hán, mặn, nhiệt độ cao, cũng như đáp ứng miễn dịch đối với các tác nhân gây bệnh. Việc phân tích scRNA-Seq đã giúp xác định được các con đường điều hòa gen và các yếu tố phiên mã có vai trò quan trọng trong việc thích nghi của thực vật với môi trường. Đặc biệt, công nghệ này cũng hỗ trợ khám phá các cơ chế biểu sinh ở mức tế bào đơn, cung cấp hiểu biết sâu hơn về cách thức kiểm soát biểu hiện gen thông qua methyl hóa DNA, biến đổi histone và cấu trúc nhiễm sắc thể. Mặc dù tiềm năng ứng dụng lớn, việc triển khai scRNA-Seq trong nghiên cứu thực vật vẫn gặp phải nhiều thách thức, đặc biệt là ở giai đoạn tách tế bào đơn từ mô thực vật do thành tế bào thực vật khá cứng và có nhiều yếu tố gây nhiễu xuất hiện trong quá trình xử lý mẫu [4]. Hơn nữa, công nghệ này cần sử dụng các phương pháp phân tích dữ liệu lớn, yêu cầu các thuật toán và công cụ tin sinh mạnh mẽ để phân tích và giải thích chính xác dữ liệu phiên mã với độ chính xác cao. Do đó, việc tiếp tục cải tiến công nghệ tách tế bào, tối ưu hóa quy trình xử lý dữ liệu, và xây dựng cơ sở dữ liệu phiên mã tế bào đơn đặc thù cho thực vật là những hướng nghiên cứu quan trọng trong tương lai.
Nguyên lý cơ bản của công nghệ phân tích hệ gen phiên mã tế bào đơn
Công nghệ scRNA-Seq là một phương pháp tiên tiến giúp phân tích biểu hiện gen ở mức độ từng tế bào riêng lẻ, thay vì thu thập dữ liệu từ toàn bộ mô như các phương pháp RNA-Seq truyền thống. Nguyên lý cơ bản của scRNA-Seq là tách từng tế bào đơn lẻ từ mẫu sinh học, sau đó ly giải tế bào để thu nhận RNA, khuếch đại RNA thành cDNA, gắn định danh phân tử duy nhất nhằm giảm nhiễu tín hiệu, cuối cùng là tiến hành giải trình tự để xác định mức độ biểu hiện của các gen trong từng tế bào. Sự phát triển của công nghệ vi lưu (droplet-based scRNA-Seq) và hệ thống mã hóa phân tử đã giúp tối ưu hóa quy trình này, cho phép thu thập dữ liệu từ hàng chục nghìn tế bào trong một thí nghiệm [5].
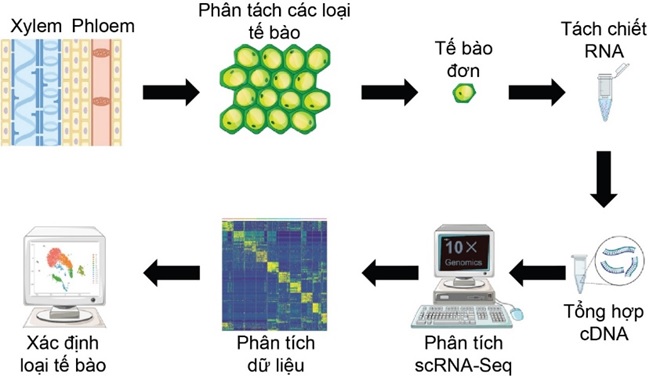
Quy trình phân tích hệ gen phiên mã tế bào đơn trong nghiên cứu thực vật.
Các bước chính trong quy trình công nghệ scRNA-Seq bao gồm một loạt thao tác kỹ thuật quan trọng, từ khâu chuẩn bị mẫu tới xử lý dữ liệu sinh học. Bước đầu tiên là tách tế bào đơn từ mô hoặc cơ quan thực vật. Đối với thực vật, bước phân lập tế bào đơn thường phức tạp hơn so với động vật do đặc điểm cấu trúc đặc trưng của tế bào thực vật ngăn cản việc tách tế bào trực tiếp bằng các phương pháp cơ học thông thường [6]. Vì vậy, để thực hiện phân tích hệ gen phiên mã ở mức độ tế bào đơn, các nhà nghiên cứu phải áp dụng một số phương pháp đặc biệt như tạo tế bào trần “sống” (loại bỏ thành tế bào nhưng vẫn duy trì sự sống và chức năng sinh lý của tế bào). Các enzyme thường được sử dụng bao gồm cellulase và pectinase, giúp phá vỡ các liên kết trong thành tế bào mà không ảnh hưởng đến cấu trúc màng tế bào và nội dung bên trong. Tuy nhiên, việc tạo tế bào trần “sống” không phải lúc nào cũng đạt hiệu quả cao và có thể gây ra những bất lợi như làm tổn thương tế bào, giảm sức sống, hoặc gây nên các phản ứng phiên mã ngoài mong muốn trong quá trình xử lý. Vì vậy, để đảm bảo chất lượng tế bào thu nhận được, các phương pháp thay thế như giải trình tự hệ gen phiên mã nhân tế bào đơn đã được phát triển và sử dụng trong nghiên cứu thực vật. Công nghệ snRNA-seq tiến hành tách các nhân tế bào riêng lẻ, tránh được những khó khăn liên quan đến thành tế bào và giảm thiểu các phản ứng phiên mã không mong muốn xảy ra trong quá trình tách tế bào. Dù vậy, cả hai phương pháp đều yêu cầu các điều kiện tối ưu hóa nghiêm ngặt, đặc biệt là điều kiện bảo quản mẫu, nồng độ và thời gian xử lý enzyme hoặc dung dịch ly giải để duy trì độ sống và toàn vẹn của vật liệu sinh học cho các phân tích tiếp theo.
Sau khi tế bào đơn được tách, RNA được chiết xuất thông qua quá trình ly giải tế bào và được chuyển đổi thành cDNA. Vì lượng RNA trong mỗi tế bào là rất nhỏ (khoảng vài picogram), bước khuếch đại cDNA là thiết yếu để thu được đủ vật liệu phục vụ giải trình tự. Các công nghệ phổ biến như Smart-seq, Smart-seq2 hoặc hệ thống 10X Genomics sử dụng các định danh phân tử duy nhất, giúp xác định được phân tử phiên mã bắt nguồn từ read, qua đó làm giảm sai lệch và tăng độ chính xác khi phân tích mức độ biểu hiện gen. Tiếp theo, các thư viện cDNA được xây dựng để đưa vào quá trình giải trình tự trên các nền tảng như Illumina hoặc hệ thống giải trình tự thế hệ mới. Các nền tảng này sẽ tạo ra lượng dữ liệu phiên mã lớn ở mức độ tế bào đơn, cung cấp thông tin chi tiết về mức độ biểu hiện của hàng nghìn gen trong từng tế bào. Việc chuẩn bị thư viện phải được tiến hành kỹ lưỡng nhằm hạn chế sự mất mát RNA cũng như giảm thiểu các lỗi kỹ thuật trong quá trình tạo thư viện, đảm bảo chất lượng dữ liệu cao nhất.
Sau khi giải trình tự, dữ liệu thô thu được xử lý ban đầu, bao gồm loại bỏ các trình tự nhiễu, sắp xếp và định vị trình tự vào hệ gen tham chiếu. Quá trình này thường sử dụng các công cụ phổ biến như Cell Ranger hoặc STARsolo để chuẩn hóa và đánh giá chất lượng dữ liệu đầu ra. Cuối cùng là giai đoạn phân tích dữ liệu, bao gồm phân tích sự biểu hiện khác biệt gen bằng các thuật toán như DESeq2 hoặc edgeR, tiếp theo là phân tích thứ nguyên dữ liệu bằng phương pháp phân tích thành phần chính hoặc thuật toán giảm chiều dữ liệu UMAP để trực quan hóa sự đa dạng phiên mã. Dữ liệu cũng được sử dụng để xây dựng bản đồ phiên mã tế bào đơn, qua đó xác định các cụm tế bào đặc thù, mô tả trạng thái phát triển, và các cơ chế điều hòa phiên mã trong điều kiện môi trường cụ thể. Những kết quả thu được từ phân tích dữ liệu này không chỉ làm sáng tỏ cơ chế sinh học cấp độ tế bào mà còn mở ra hướng nghiên cứu mới phục vụ phát triển các giống cây trồng chống chịu tốt hơn với điều kiện bất lợi từ môi trường.
Ưu điểm của công nghệ trong nghiên cứu cây trồng
Công nghệ scRNA-Seq có nhiều ưu điểm nổi bật so với các phương pháp phân tích hệ gen phiên mã truyền thống, đặc biệt trong nghiên cứu thực vật. Thứ nhất, scRNA-Seq cung cấp độ phân giải cao ở mức tế bào đơn, cho phép nhận diện và mô tả rõ ràng tính đa dạng của các tế bào trong cùng một mô hoặc cơ quan thực vật. Các phương pháp truyền thống thường chỉ phân tích mẫu ở cấp độ toàn bộ mô, làm mất đi thông tin đặc hiệu phiên mã của từng loại tế bào, nhất là các tế bào hiếm hoặc tế bào chuyên biệt. Ví dụ, công nghệ scRNA-Seq được sử dụng để phân tích bản đồ phiên mã với hơn 20.000 tế bào từ vùng rễ của hai giống lúa Nipponbare (Japonica) và 93-11 (Indica). Kết quả cho thấy rõ tính đa dạng của hệ gen phiên mã với 8 loại tế bào chính tại biểu bì, lông hút, vỏ rễ, nội bì, mạch dẫn, tế bào xylem và chóp rễ liên quan đến cơ chế thích nghi của cây lúa với sự thay đổi của môi trường [7].
Một ưu điểm khác của scRNA-Seq là khả năng phát hiện các loại tế bào hiếm và xác định vai trò sinh học cụ thể của chúng trong điều kiện phát triển bình thường cũng như khi đáp ứng với các yếu tố bất lợi. Các phương pháp truyền thống thu thập RNA từ toàn bộ mô sẽ làm mờ đi tín hiệu biểu hiện gen của những loại tế bào chiếm tỷ lệ thấp, dẫn tới sự bỏ sót thông tin phiên mã quan trọng. Ngược lại, scRNA-Seq giúp xác định rõ các tế bào này và mô tả một cách chi tiết sự biểu hiện phiên mã đặc trưng, làm cơ sở cho nghiên cứu chuyên sâu về chức năng và cơ chế điều hòa gen. Ví dụ, bằng cách lập bản đồ hệ gen phiên mã chi tiết từ 25.166 tế bào đơn của Populus trichocarpa, một số tế bào hiếm quan trọng trong quá trình biệt hóa xylem, bao gồm tế bào trung gian hình thoi và tế bào tổ chức tia đã được phát hiện [8]. Những tế bào này có tỷ lệ thấp trong tổng số tế bào xylem, khiến chúng khó phát hiện bằng các phương pháp truyền thống. Tuy nhiên, nhờ scRNA-Seq, nghiên cứu đã xác định chính xác các nhóm tế bào này và làm sáng tỏ vai trò của chúng trong sự phát triển của xylem [8].
Công nghệ scRNA-Seq không chỉ giúp xác định sự khác biệt của hệ gen phiên mã giữa các loại tế bào mà còn có khả năng theo dõi động lực học phiên mã theo thời gian. Bằng cách sử dụng phân tích quỹ đạo phát triển tế bào, scRNA-Seq giúp xác định trình tự phân hóa tế bào, đánh giá trạng thái phát triển của tế bào tại từng giai đoạn, đồng thời xây dựng bản đồ hệ gen phiên mã theo thời gian. Điều này có ý nghĩa quan trọng trong việc hiểu rõ hơn cơ chế điều hòa phiên mã trong sự sinh trưởng và đáp ứng của tế bào thực vật trước các yếu tố môi trường. Bằng phương pháp phân tích quỹ đạo phát triển tế bào, nghiên cứu gần đây đã chứng minh rằng sự biệt hóa tế bào xylem không diễn ra đồng nhất mà có sự khác biệt theo từng loài [8]. Ví dụ, P. trichocarpa và Eucalyptus grandis có quỹ đạo biệt hóa xylem theo từng giai đoạn tương tự nhau, trong khi Liriodendron chinense có con đường biệt hóa xlylem khác biệt, phản ánh sự đa dạng tiến hóa của mô xylem trong thực vật hạt kín [8]. Hơn nữa, khi so sánh với cây mô hình hai lá mầm Arabidopsis thaliana, kết quả đã chỉ ra rằng các tế bào xylem trong cây thân gỗ có hệ thống biệt hóa phức tạp và kéo dài hơn, cho phép cây phát triển hệ mạch dày đặc để vận chuyển nước hiệu quả hơn [8]. Bằng cách xây dựng bản đồ hệ gen phiên mã theo thời gian, scRNA-Seq giúp xác định các gen chủ đạo điều khiển quá trình biệt hóa tế bào xylem, chẳng hạn như nhóm gen HD-ZIP III, có vai trò kiểm soát trạng thái tế bào tổ chức, đóng vai trò chính trong việc kích hoạt biệt hóa yếu tố mạch [8]. Những thông tin này có giá trị ứng dụng trong nghiên cứu cải tiến giống cây trồng, đặc biệt là trong việc điều chỉnh cấu trúc gỗ và khả năng thích nghi với môi trường của cây trồng [1, 3].
Công nghệ scRNA-Seq không chỉ giúp phân tích sự khác biệt về biểu hiện gen giữa các tế bào mà còn có thể tích hợp với các công nghệ tiên tiến khác như giải trình tự hệ gen phiên mã theo không gian và đa -omic tế bào đơn để cung cấp cái nhìn toàn diện về sự điều hòa gen trong không gian và thời gian [9]. Sự tích hợp này giúp kết nối dữ liệu hệ gen phiên mã với thông tin không gian của tế bào, cấu trúc nhiễm sắc thể, và các biến đổi biểu sinh, mở ra hướng nghiên cứu mới về mạng lưới điều hòa gen và cơ chế phát triển thực vật mà các phương pháp phân tích hệ gen phiên mã truyền thống không thể thực hiện được. Như vậy, với những ưu điểm vượt trội về độ phân giải, khả năng phát hiện các tế bào hiếm, phân tích động lực học phiên mã và tích hợp đa dữ liệu, công nghệ scRNA-Seq đang trở thành công cụ quan trọng và mạnh mẽ trong nghiên cứu sinh học thực vật hiện đại.
Thay lời kết
Việc ứng dụng scRNA-Seq trong nghiên cứu thực vật mang lại nhiều tiềm năng nhưng cũng đi kèm với một số thách thức. Các nhà nghiên cứu cần lưu ý đến quy trình tách tế bào, xử lý RNA, lựa chọn công nghệ phù hợp và tích hợp dữ liệu với các phương pháp khác để đạt được kết quả chính xác nhất. Bên cạnh đó, việc mở rộng áp dụng trong nông nghiệp cần cân nhắc rất thấu đáo về chi phí và khả năng phân tích dữ liệu.
RNA trong scRNA-Seq dễ phân hủy, cần bảo quản kỹ và xử lý trong môi trường hạn chế RNase. Do RNA từ một tế bào đơn quá ít để phân tích trực tiếp, phải khuếch đại nhưng dễ gây sai số, được khắc phục bằng công nghệ gắn định danh phân tử. Phân tích scRNA-Seq phụ thuộc vào hệ gen tham chiếu, nhưng nhiều loài cây trồng chưa có bộ gen đầy đủ, cần cơ sở dữ liệu phiên mã chính xác hơn. Để hiểu rõ sự phát triển tế bào thực vật, scRNA-Seq cần tích hợp với các công nghệ khác, đòi hỏi thuật toán mạnh để đồng bộ dữ liệu.
Mặc dù scRNA-Seq là một công nghệ tiên tiến, nhưng chi phí thực hiện vẫn còn cao, đặc biệt là đối với các nghiên cứu trên hàng nghìn đến hàng chục nghìn tế bào. Ngoài ra, dữ liệu scRNA-Seq rất lớn và phức tạp, đòi hỏi công cụ phân tích tin sinh học mạnh mẽ như Seurat, Scanpy và Monocle để tiền xử lý, lọc nhiễu và phân cụm tế bào. Do đó, các nhóm nghiên cứu cần phải có kỹ năng về tin sinh học hoặc hợp tác với các chuyên gia trong lĩnh vực này để phân tích dữ liệu chính xác.
TÀI LIỆU THAM KHẢO
[1] M. Luo, Y. Cao, J. Hong (2025), "Opportunities and challenges in the application of single-cell transcriptomics in plant tissue research", Physiology and Molecular Biology of Plants, 31(2), pp.199-209, DOI: 10.1007/s12298-025-01558-6.
[2] B. Cole, D. Bergmann, C.E.B. Haas, et al. (2021), "Plant single-cell solutions for energy and the environment", Communications Biology, 4(1), DOI: 10.1038/s42003-021-02477-4.
[3] D. Zheng, J. Xu, Y. Lu, et al. (2023), "Recent progresses in plant single-cell transcriptomics", Crop Design, 2(2), DOI: 10.1016/j.cropd.2023.100041.
[4] C. Chen, Y. Ge, L. Lu (2023), "Opportunities and challenges in the application of single-cell and spatial transcriptomics in plants", Frontiers in Plant Science, 14, DOI: 10.3389/fpls.2023.1185377.
[5] R. Shaw, X. Tian, J. Xu (2021), "Single-cell transcriptome analysis in plants: Advances and challenges", Molecular Plant, 14(1), pp.115-126, DOI: 10.1016/j.molp.2020.10.012.
[6] Y. Sun, J. Sun, C. Lin, et al. (2024), "Single-cell transcriptomics applied in plants", Cells, 13(18), DOI: 10.3390/cells13181561.
[7] Q. Liu, Z. Liang, D. Feng, et al. (2021), "Transcriptional landscape of rice roots at the single-cell resolution", Molecular Plant, 14(3), pp.384-394, DOI: 10.1016/j.molp.2020.12.01
[8] C.C. Tung, S.C. Kuo, C.L. Yang, et al. (2023), "Single-cell transcriptomics unveils xylem cell development and evolution", Genome Biology, 24(1), DOI: 10.1186/s13059-022-02845-1.
[9] K. Adema, M.A. Schon, M.D. Nodine, et al. (2024), "Lost in space: What single-cell RNA sequencing cannot tell you", Trends in Plant Science, 29(9), pp.1018-1028, DOI: 10.1016/j.tplants.2024.03.010.